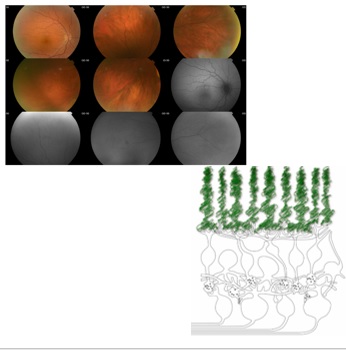
Enfermedades Oculogenéticas (EO)
Las Distrofias hereditarias de la Retina (DR) son responsables del 5% de la ceguera en el mundo occidental, siendo la causa más común de pérdida de visión en niños y adultos jóvenes.
En conjunto afectan a 1 de cada 3000 a 4000 personas. Afectan de modo primario a los fotoreceptores y células del epitelio pigmentario de la retina (EPR), produciendo degeneración progresiva y finalmente apoptosis.
Clínicamente se dividen en:
i) formas periféricas, con afectación inicial y predominante de los bastones (ej: Retinosis Pigmentaria (RP), Amaurosis congénita de Leber (LCA)) y,
ii) formas centrales, con degeneración de los conos en su inicio (Distrofias maculares (DM), distrofias de conos), pudiendo afectarse secundariamente los bastones, en las distrofias de conos-bastones (DCB).
El 20-30% de los pacientes presenta además síntomas extraoculares (DR sindrómicas (DRS)).
Las DR presentan también una gran heterogeneidad genética, con cerca de 293 loci y 256 genes descritos (https://sph.uth.tmc.edu/Retnet/ ) que codifican para proteínas con distintas funciones (desarrollo de la retina, fototransducción, ciclo visual, transcripción, transporte nuclear, fagocitosis, splicing y estructurales, incluyendo las ciliares) en los fotorreceptores o las células del EPR.
) que codifican para proteínas con distintas funciones (desarrollo de la retina, fototransducción, ciclo visual, transcripción, transporte nuclear, fagocitosis, splicing y estructurales, incluyendo las ciliares) en los fotorreceptores o las células del EPR.
Todos ellos sólo explican entre el 40-60% de los casos. El equipo de la FJD tiene una larga experiencia en el estudio de las DR desde hace más de 27 años, habiendo organizado cohortes y caracterizando varios nuevos genes.
Además, estudia otras enfermedades oculogenéticas, tanto del desarrollo (malformaciones) junto con los grupos del RyC y de la Paz, así como neurodegenerativas (atrofia del nervio óptico) junto con el grupo UAM y del 12Oct.