Enfermedades Mitocondriales (EMT)
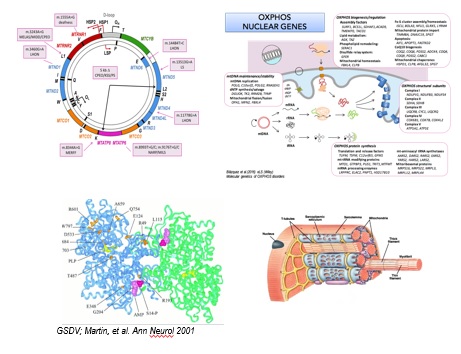
Las enfermedades mitocondriales (EMT) aunque individualmente son raras, en su conjunto, constituyen un amplio grupo de enfermedades genéticas cuyo nexo de unión es un defecto en el sistema de fosforilación oxidativa (OXPHOS).
El sistema OXPHOS se encarga de sintetizar la mayor parte del ATP celular y se localiza en la membrana interna mitocondrial.
La biogénesis de este sistema requiere la expresión coordinada de dos genomas, el mitocondrial y el nuclear; mutaciones en ambos pueden dar lugar a la enfermedad. En general, las EMT son un grupo de enfermedades genéticas muy heterogéneo, caracterizadas por una gran variedad de presentaciones clínicas, con una importante presencia de síndromes neurológicos y causantes de una tasa significativa de morbidez y mortalidad, principalmente durante la infancia.
Suelen afectar preferentemente a los tejidos con una alta demanda energética como cerebro, músculo esquelético, corazón y sistema endocrino.
Las enfermedades OXPHOS normalmente cursan con un deterioro progresivo, lo que provoca discapacidad, y en algunos casos, muerte prematura, frecuentemente debida a defectos en la conducción cardíaca o a la presencia de convulsiones.
Aunque se ha avanzado mucho con las nuevas tecnologías genómicas se desconoce todavía el defecto genético en muchas de estas patologías, por lo que la caracterización genética en definitiva representa un reto para la mejora terapéutica de los pacientes.
Los grupos del i12O, CBMSO y UAM tienen una amplia experiencia en la caracterización bioquímica y molecular de todas estas enfermedades neurometabólicas incluyendo estudios genéticos basados en NGS, la caracterización de las células mutantes, el modelado de la patología en otros sistemas biológicos y la aplicación de terapias variante-específicas.